



















科研人员开发出可解释AI模型精准鉴定细胞谱系特征基因
2026/05/29
 88
88细胞类型的精准识别是单细胞转录组学分析的基础,而发现细胞类型特异性的标记基因是实现这一目标的关键。传统方法多依赖统计阈值或聚类启发式策略,易受数据噪声、注释偏差及基因高表达但非特异性等问题的干扰。
近日,中国科学院广州生物医药与健康研究院科研团队基于可解释神经网络框架,提出了scMarkerGene模型。该模型通过构建“贡献分数矩阵”,将神经网络模型中每个基因对细胞类型判别的影响量化为可解释的贡献值,并结合集成学习与特异性过滤策略,实现了对不同物种、不同测序技术、不同细胞群体规模及高稀疏性数据的稳健标记基因识别。
scMarkerGene的工作流程主要包括两个步骤。第一步是贡献分数计算。基于多层感知机构建分类模型,通过集成多个超参数扰动训练得到的模型,利用DeepLIFT解释方法计算每个基因对细胞类型判别的贡献分数,并经过统计检验筛选出候选标记基因。第二步是特异性筛选与重排序。在候选基因基础上,结合基因的均值表达、中位数表达及检出率,构建“marker评分”,并与轮廓系数加权后对基因进行重排序,最终输出高特异性的细胞类型标记基因。
研究团队在10个公开的单细胞RNA测序数据集上对scMarkerGene进行了系统评估,涵盖拟南芥、果蝇、小鼠、人类等多个物种及多种测序平台。结果表明,这一模型在log2FC、标准化Z-score等指标上均优于scanpy、scMAGs、SMaSH、scVI等现有方法。在模拟数据实验中,scMarkerGene识别高特异性标记基因,明显领先其他方法,并可有效滤除非特异性基因。在引入不同比例的随机丢失噪声后,scMarkerGene依然保持高鲁棒性,而同类方法SMaSH的性能则明显下降。在骨类器官数据集中,scMarkerGene在粗粒度与细粒度细胞类型上均能稳定识别高特异性标记基因,尤其在样本量不足100个细胞的小群体中仍保持最高log2FC,展现出对罕见细胞群体的强大适应性。
团队进一步在空间转录组与拟时间序列分析中发现,scMarkerGene识别出的标记基因在10X Visium小鼠脑组织及人黑色素瘤数据中均展现出清晰的空间定位特征;在BEELINE基准数据中,其在不同离散时间状态下预测的标记基因也均取得较高的log2FC值。
scMarkerGene区别传统方法依赖表达均值差异检验的方式,以判别函数为核心,以贡献分数为统一度量,建立起基因贡献分数与分类决策边界敏感度之间的数学联系,推动标记基因筛选从“统计描述”走向“机制解析”,为从复杂单细胞数据中解析细胞身份提供了可靠方法。
相关研究成果发表在Briefings in Bioinformatics上。研究工作得到国家重点研发计划等的支持。
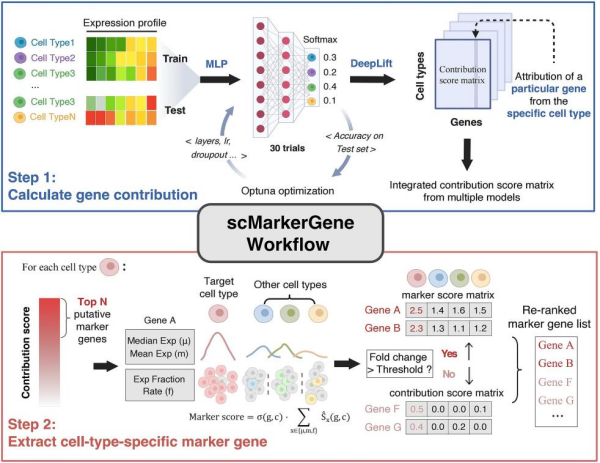
scMarkerGene工作流程
文章来源广州生物医药与健康研究院,分享只为学术交流,如涉及侵权问题请联系我们,我们将及时修改或删除。







-
2026年第九届可再生能源和电力工程 443

-
2026年第二届计算机视觉与机器学习 1817
-
2026年6月优质国际学术会议推荐 2215
-
2026年无人机遥感与人工智能国际会 240
-
2026资源、化学化工与应用材料国际 3575
-
2026年图像处理与数字创意设计国际 3383
-
2026年机械工程,新能源与电气技术 7893
-
2026年材料科学、低碳技术与动力工 3538
-
2026海洋科学、探测技术与航海工程 07-27

-
2026建筑规划、环境工程与城市交通 07-27
-
2026医学材料、仿生工程与生物制造 07-27
-
2026机器视觉、模拟仿真与大模型技 07-27
-
2026核物理、量子信息学与电声技术 07-27
-
2026年电子工程、通信系统与信号处 07-27
-
2026年人工智能、艺术疗愈与心理健 07-27

































-
2026 JCR影响因子正式发布896
-
中国科协发布2025年《重要学术1445
-
2026年新锐分区(原中科院期刊7742
-
2025年两院院士增选有效候选人5840
-
好学术:科研网址导航|学术头条分7617
-
2025年国际期刊预警名单发布!7848
-
2025年中科院期刊分区表重磅发27775
-
吉林大学校长张希:学术会议中的提8895
-
北京大学生命科学学院焦雨铃团队与07-26
-
清华大学计算机系孙富春团队在具身07-26
-
研究揭示寄主水力性状限制桑寄生抗07-26
-
同域栎树气候适应性策略研究取得进07-26
-
上海交大Bio-X研究院平勇与合07-26
-
上海交大中英国际低碳学院纪亚副教07-26
-
上海交大集成电路学院吴泳澎教授、07-26
-
清华大学出版社有限公司 21435

-
内蒙古集宁师范高等专科学校生化系 18541
-
HKSME 23331
-
北京全卫联合医学科学研究院 2566
-
上海商图信息咨询有限公司 23673
-
天津市国土资源与房屋职业学院 18533
-
江苏扬子会展服务有限公司 4393
-
西北大学 24575
-
世界医联(北京)国际中医药研究院 23773
-
北京师范大学 23552
-
昆明市兴达会议有限公司 18576
-
华汉广告公司 21494
-
解放军理工大学工程兵工程学院 21775
-
北京万邦会展有限公司 21480
-
fdcv 23357
-
巨燈照明有限公司 21416
-
华中农业大学文法学院 23594
-
InnovationEnterp 21666
-
VREAFEW 24540
-
海纳集团 24063



